PHOGEMON PROJECT 公式マニュアル
Phosphorylation and guanine-nucleotide exchange monitors (PHOGEMON)
京都大学生命科学研究科生体制御学・医学研究科病態生物医学
編著者: 中野大勢、松田道行(matsudam.michiyuki.2c (atto) kyoto-u.ac.jp)。
改訂の記録:
第1版 平成13年4月18日
第10版 平成29年3月31日
(詳細なデータ等は英語版ホームページに入れるように努力してますのでそちらをご覧ください。)
Raichuの動作原理 By Takashi Ohki
序文
21世紀生物学の目標の一つがコンピューター上で動くインシリコ細胞の作成にあることは衆目の一致するところでしょう。わたしたちが夢見ているのは、「細胞のがん化過程を再現できるインシリコの細胞の創生」です。それを使えば、もっとも効果的な抗がん剤の標的物質を見つけることができるでしょう。さらには、個人個人の癌細胞が有している変異に応じて、がん化プロセスを再現することもできます。そうれば、個人のがん細胞の特性に応じて、どの分子を標的にした薬剤が必要か、どの薬剤の組み合わせがもっとも有効かということを、コンピューター上で予測できるようになるでしょう。すなわち、個々の癌細胞にもっとも適した治療法を解答してくれるインシリコ細胞を作ること、が私たちの目的なのです。しかし、現在の細胞生物学が有している情報は、細胞のシミュレーションを行うには全く足りません。それは、現代生物学の隆盛を築いた生化学・分子生物学が、「細胞を材料として均一化した後に分子を解析する」という手法に立脚しているために、分子が細胞内のいつでどこ活性化されるか?という時空間情報を得ることができないからです。私達の目的は、この情報伝達の時空間パラメータを得る技術を開発し、それをもとに様々な細胞の働きを分子レベルで解明することです。私たちはまず細胞内情報伝達の主役であるリン酸化反応とグアニンヌクレオチド交換反応が起こる様子の画像化に取り組みました: 名づけてPhosphorylation and guanine-nucleotide exchange monitoring project (PHOGEMONプロジェクト)。このプロジェクトは研究室が東京の国立国際医療センターにある時に開始しましたが、その後、研究室は大阪に移り、さらに平成18年からは任天堂本社のある京都に動くことになりました (いうまでもありませんが、このプロジェクトと世界的に有名なポケモンとは何の関係もありません)。
ScienceとTechinologyは不可分です。どの分野の研究が伸びるかはどんな技術が開発されつつあるかに依存しているのは自明です。本文ではわたしたちの作った道具で自分の研究を新しい側面から見てみたいという方や、自分でもプローブを作ってみたいという方のために、わたしたちの研究の内容をご紹介します。なお、技術上の詳細な情報は英語版に書いてありますので、さらに詳しく調べたい方はそちらを参照してください。最後に、われわれのグループは光学やシステム生物学の専門家では決してありませんが、いろんなところに首を突っ込んで研究を広げていきたいと思っております。本文には、多々間違いもあると思いますが、遠慮なくご指摘ください。
生体イメージングの始まり(2017年追記):
これまで、当研究室の興味は細胞内情報伝達系の時空間変化でした。最近は、情報伝達の組織レベルでの時空間変化に興味がシフトしつつあります。それは、培養皿の細胞は生命を理解するためのあくまでも通過点に過ぎず、真理は生きた生物の多様な細胞から構成させる組織で研究しないとわからないからです。実際の組織の中は培養系とは異なり、多くの細胞がコミュニケーションをとりながら生命の維持を行っているのです。このような研究が可能となったのは二つのブレークスルーのおかげです。ひとつは、トランスポゾンを使って高感度FRETバイオセンサーをマウスに安定して発現させる技術が確立したこと、二つは二光子顕微鏡を用いてFRETイメージングを各種臓器で行う技術が確立されたことです。現在、様々なマウスを使って、生きた組織で情報伝達分子がどう変化し、それが生体にどう影響を及ぼすのかを研究しています。
序論
細胞内情報伝達研究の新しい流れ:
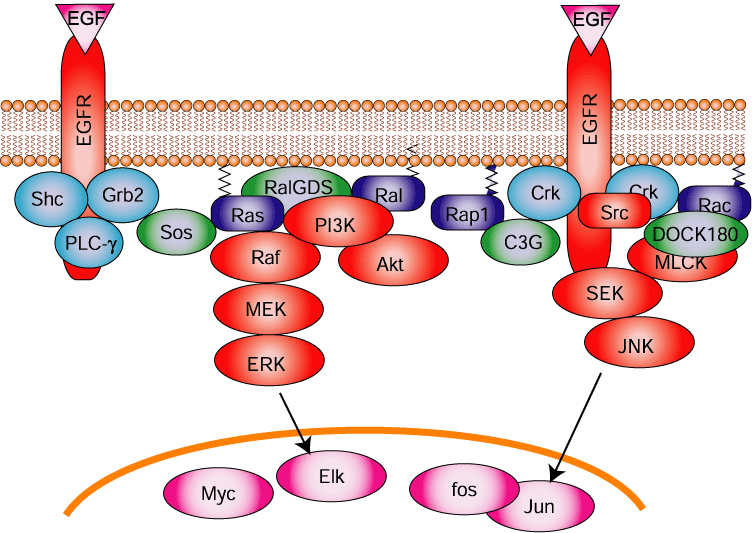
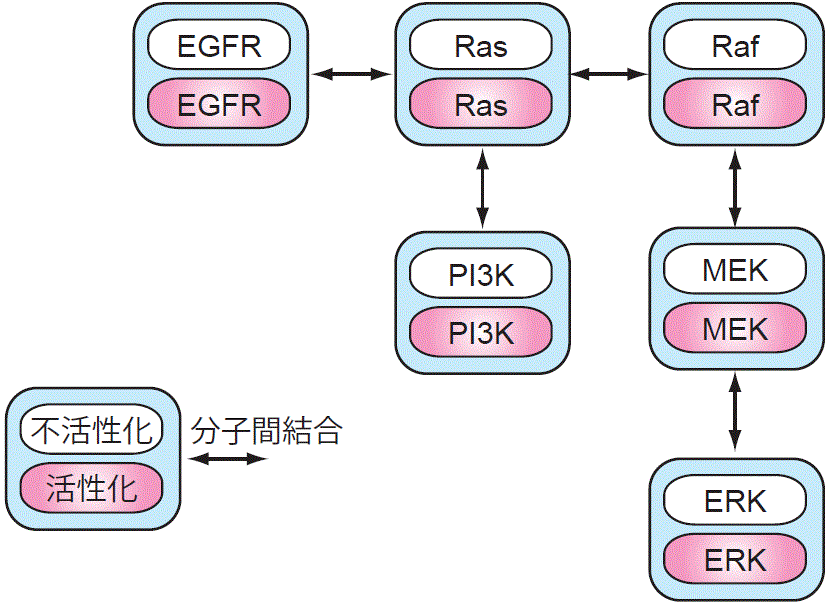
ヒトゲノムの全配列が決定され、細胞内情報伝達に登場する役者は全て出揃ったと考えられています。ですから、現在の情報伝達研究は、一つの細胞内に存在する数万の分子から構成されるネットワークをどうやって再構築するかという点に絞られています。その最有力な手法として用いられているのがタンパク質ータンパク質間の相互作用(特異的結合)を網羅的に解析するという手法です。そのようなタンパク質間相互作用を指標としたネットワークとしては例えば図3.1のようなモデルが使われています(細胞増殖シグナル系)。しかしながら、このようなモデルは細胞内シグナルがどのように伝播していくかという時空間情報を全く無視したもので、実際の細胞の情報伝達とは依然大きな隔たりがあります。この問題に立ち向かうべく、『生きた細胞・組織・動物』で情報伝達を研究しようという動きが始まっています。この動きは蛍光タンパク質の発見なくしては語れないので、まず緑色蛍光タンパク質の話からスタートします。
緑色蛍光タンパク質:
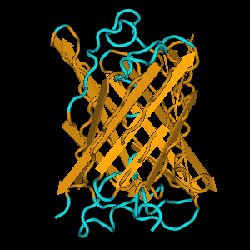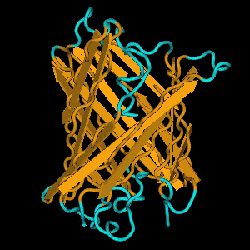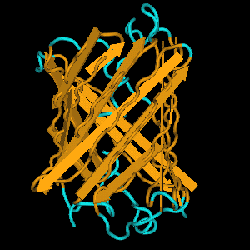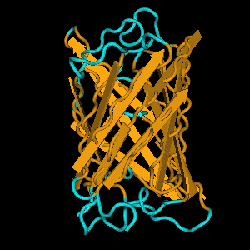
GFP(Green Fluorescent Protein)はオワンクラゲの作る緑色蛍光タンパク質として単離され、いまや、細胞生物学にとって無くてはならないツールとなっています。2008年に下村博士らにノーベル賞が授与されたのは当然のことでしょう。その後、オワンクラゲ以外の生物からも次々に蛍光タンパク質が単離され、青から近赤外まで実に多くの色の蛍光タンパク質が使えるようになっています。また、結晶構造やランダム変異を用いる方法などを駆使して改良が進められてきました。当初は目的とするタンパク質のマーカーとして使うだけでしたが、その後、様々な細胞内事象のモニターとしての用途、さらには超解像顕微鏡への応用など驚くべき発展を見せています。詳細は(文献1-2、文献45)を参照してください。
PHOGEMON総論
図4.1.1
FRETとは:
FRETはFluorescence Resonance Energy Transfer (蛍光共鳴エネルギー移動)の略であると記載している論文が生物系雑誌には多かったのですが、最近は、発見者の名前に由来するFörster Resonance Energy Transfer (フェルスター共鳴エネルギー移動)の略とする論文が増えています。詳しい解説はこちらをごらんください。われわれが開発しているバイオセンサー(プローブともいいます)は、緑色蛍光タンパク質(GFP)のシアン色変異体であるCFPから、黄色変異体であるYFPへのFRETを利用しています(図4.1.1)。CFPをドナー、YFPをアクセプターと呼びますが、ドナーとアクセプターの組み合わせはCFP/YFP以外にもたくさんあります。また、GFP変異体はそれぞれに特性が異なるので、それらを良く理解する必要があります。我々が使用しているYFPやCFPにも、いろんなversionがあって、目的にあったものを使っています(YFP, CFP)(文献12-17)。
図4.2.0
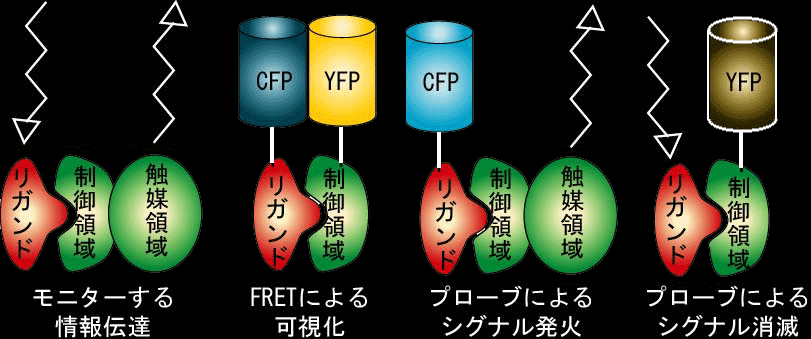
シグナル伝達ネットワークの動作原理:
シグナル伝達ネットワークの基本原理は、シグナル伝達分子の活性変化とシグナル伝達分子間の特異的相互作用です。個々のシグナル伝達分子はスイッチと考えられ、ONとOFFの状態があります(図の赤と白の分子ペア)。ONになると次のシグナル伝達分子に結合し、シグナルを伝播します(図中の矢印)。ですから、シグナル伝播を観るためには、スイッチのON/OFF状態あるいはシグナル伝達分子間の結合を可視化すればいいのです。
図4.2.1
1分子FRETと2分子FRET:
CFPとYFPとがFRETのドナーとアクセプターとなることを利用すれば、2分子の会合状態を生きた細胞でモニターすることができます。図4.2.1で示すように、会合状態を調べたい2分子をそれぞれCFPとYFPの融合タンパク質として発現すれば、会合した時にだけFRETを観察することが可能です。また、この2分子を融合させて1分子にすることも可能で、私達の使っているPHOGEMONはこの1分子FRETのシステムを採用しています(図4.2.2)。 また、1分子FRETと2分子FRETの中間ともいうべき、1.5分子FRETシステムもあります(図4.2.2a)。以下にそれらの得失を簡単に紹介します。
図4.2.3
2分子FRETの特徴 (図4.2.1, 4.2.3):
2分子FRETの系を構築するのは比較的簡単です。この図の赤い分子と緑の分子が刺激依存性に結合するとしましょう。まず、この赤と緑の分子をそれぞれCFP(青)とYFP(黄)に融合させて発現させます。すると、赤分子と緑分子がくっつくとFRETが起きてYFPが光るシステムができます。この2分子FRETの系の最大の欠点は、シグナルノイズ比(S/N比)が低い点です。これはCFPの蛍光特性に大きく依存していますが、ほかの蛍光タンパク質でも必ず起こる問題です。この問題は次節で詳しく触れます。さらなる問題点として、内在性の情報伝達を活性化してしまったり、逆に生理的情報伝達を阻害してしまったりという欠点が考えられます(図4.2.3)。
図4.2.2
1分子FRETの特徴 (図4.2.2):
2分子FRETの系を一つにくっつけただけのものです。この系は、FRETに参加するCFPの割合が高いのでS/N比が2分子FRETよりも格段にいいのが特徴です。また、内在性の情報伝達を阻害する可能性も低く抑えられます。ここで大事なことは、この図で緑と赤の色で示した部分は、分子間会合をする異なる分子であってもいいし、分子内会合を司る同一分子の二つのドメインであってもよい、ということです。すなわち、後者の場合、このプローブは、ある一つのタンパク質の構造変化を捉えているといえます。ここで、「すべてのタンパク質の機能的変化は、その構造変化を伴う」ということを想起してください。このことに気がつけば、この1分子FRET法は、理論上は「あらゆるタンパク質の活性変化を検出できる」ということがいえるでしょう。この系の難点は次の3つに集約されます。①プローブが大きくなるので、プラスミドの作成が面倒である。②大腸菌等で作ったタンパク質は不溶性になることが多く、精製標品を使った解析が困難である。③FRETが起きるための空間的配置を試行錯誤で見つけなければならない。とはいえ、できてしまえばこっちのもので、ユーザーに徹する方は、このタイプのプローブをもらうのが望ましいでしょう。現在知られているGFPを利用した1分子FRETプローブをまとめました。
図4.2.2a
1.5分子FRETシステム (図4.2.2a:
この系は、1分子FRETに属するものですが、標的分子がプローブ外から来るところに特徴があります(図の緑のボール)。つまり、内在性の緑分子がプローブ内の赤分子に結合するとFRETが減少するシステムです。この系は、GFPがダイマーを作りやすいという蛍光があるのを逆手に取っています。この系はプローブ作成が楽であるという2分子FRETの系と、S/N比が高いという1分子FRETの系の両方の利点を有しています。ただし、うまくいかなかったときに、改良の余地が少なこと、プローブの濃度が内在性の標的分子より少なくないと、①S/N比があがらない、②内在性のシグナルを止める、という欠点があり、必然的にあまり明るくはできません。
図4.3.1
FRET効率の測定:
FRET効率の測定は、下記の3つが主に用いられます。
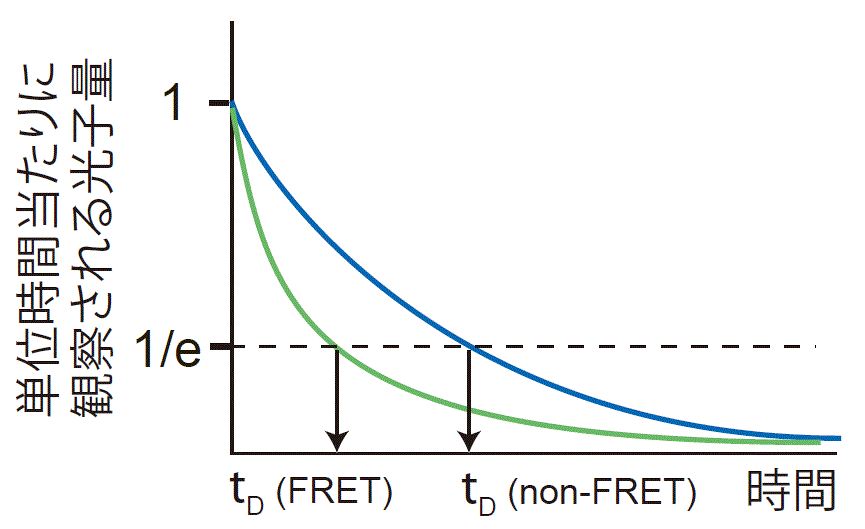
Fluorescent Lifetime Measurement (蛍光寿命測定法):
この方法がFRETの定義にもっとも忠実な方法で、定量性も高いものです。蛍光寿命は、パルス上の光を照射し、その後の蛍光減弱を光子をカウントすることにより調べる時間領域測定法(Time domain)(図4.3.1)と、入射光のパワーと検出器の感度に変調を与えることで位相のずれを検出する方法(Phase domain)とがあります。いずれも特殊な検出器が必要となります。レーザーを操作することで顕微鏡画像を得ることもでき、これをFluorescence Lifetime Imaging Microscopy (FLIM)と呼んでいます。 蛍光寿命顕微鏡の使用法はこちらをご覧ください。
図4.3.2
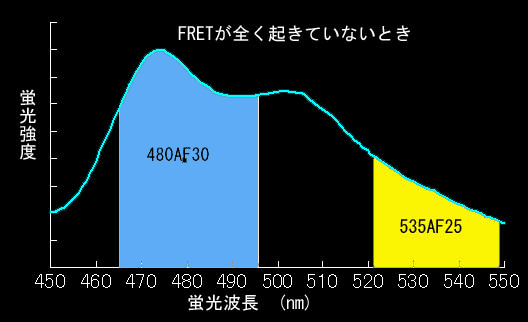
図4.3.3
Acceptor Bleaching (アクセプターブリーチング):
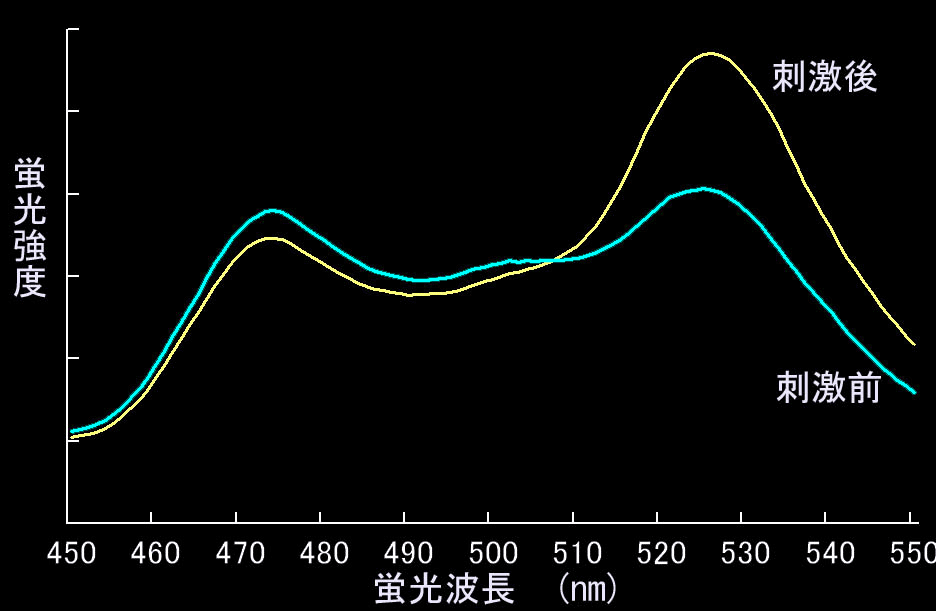
既述の通り、蛍光寿命は蛍光強度とは比例しますが、蛍光強度は分子数にも依存しますので、そのままではFRETの評価には使えません。そこで、アクセプター分子に強い光を当てて褪色を誘導し、それによるドナーの蛍光が増強することによって、”FRETがあった”ことを簡便に、ただし、定性的にのみ証明することができます。図4.3.3から図4.3.2の状態へ変化すればFRETがあったということになります。
Sensitized FRET (感応性FRET) :
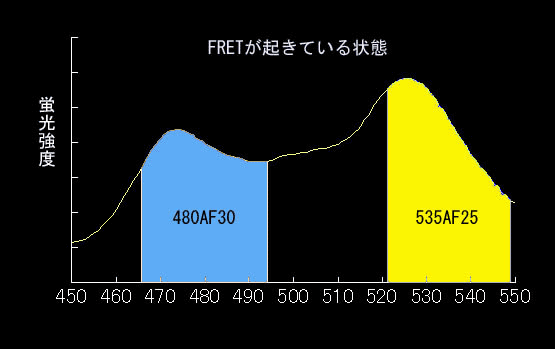
FRETはドナーの蛍光寿命減少が第一の定義ですが、多くの場合アクセプターからの蛍光が観察されます。これをSensitized FRET(感応性FRET:この訳が適当かどうかは不明)と呼んでいます。これがもっとも一般的に用いられる方法ですので、以下、CFPとYFPを例にとり説明します。まず433 nmでCFPを励起したときのCFPの蛍光プロフィールを見てください(図4.3.2)。かなり広い波長域の蛍光を発します。ですから、実際のイメージングにおいては、CFPの蛍光強度は480±30 nmのバンドパスフィルターで取り込み(図4.3.2の青い部分)、YFPの蛍光強度は535±25のバンドパスフィルターを使って測定します(図4.3.2の黄色い部分)ので、FRETが全くない場合でも、YFPのチャンネルに光が検出されます(この漏れこみをbleedthroughといいます)。ですから、YFPチャンネルで蛍光を測定するだけではFRETが起きているかどうか知るすべはありません。そこでFRET効率を調べるためにYFP/CFPの値を使います。このように二つの比を取る測定法をratiometryといい、データの規格化としてよく使う方法です。FRETがまったく起きていないときは、YFP/CFP値は約0.2くらいになります。一方、FRETが起きると図4.3.3のようにCFPの蛍光が減ってYFPの蛍光が現れます(図4.3.3)。
図4.3.4

図4.3.5
例をあげて説明しましょう(図4.3.5)。情報伝達分子Aが活性化されてBに結合する系を考えます。このAとBの結合をFRETで検出するために、情報伝達分子AにCFPを、BにYFPを融合させて系を構築します。図の下に書いてある数字は、475 nmおよび530 nmの光がどれくらい検出できるかを、それぞれの状態について、書いてあります。刺激後、すなわちFRETが誘導されたときの数字を赤で書いてあります。顕微鏡では個々の分子を識別せずにこれらの分子を丸ごと捉えますので、刺激前はYFP/CFP=1.4/7=0.2, 刺激後は1.7/6.5=0.26という数字が出てきます。すなわち、この系で1割強の分子が会合したとしても、シグナル(YFP/CFP)のゲインは30%にとどまります。通常、増殖刺激などにおいて、シグナル伝播に関わる分子は1割以下と言われていますから、実際には10%のゲインはなかなか稼げないでしょう。もちろん十分に明るい条件で実験すれば10%のゲインでも十分なのですが、実際にはこの程度の、ゲインでは十分とはいえません。一方、1分子FRETの場合には、非会合時にもFRETは起きているという問題があります。ですから、通常使用されているプローブでは、刺激の有無での蛍光強度比の変化は50%を超えれば上出来だ、ということを認識する必要があります(図4.3.5)。
PHOGEMONの基本構造:
図4.4.1
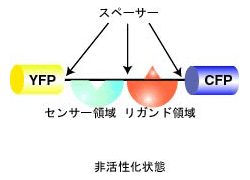
図4.4.2
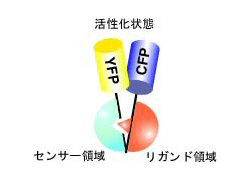
PHOGEMONの基本構造を説明します。PHOGEMONは、N末端にYFP、C末端にCFPを有し、その間にセンサー領域とリガンド領域を有するのが基本構造です(図4.4.1)。 センサー領域とは、細胞内の刺激に応じて構造が変化する部分のことです。例えば、Gタンパク質(GTPが結合すると構造が変化する)や様々な酵素のリン酸化部位がそれに相当します。リガンド領域とは、これらの構造変化を認識するタンパク質質のドメインで、Gタンパク質に対しては標的分子(エフェクターとも呼ばれます)、リン酸化部位に対してはSH2ドメイン(リン酸化チロシンを認識する)などがそれに相当します。PHOGEMONはセンサー領域の構造が変化し、リガンド領域と結合すると、CFPとYFPとが近くなり、FRETが起きるように設計します(図4.4.2)。以下、簡便のために、これらを非活性化状態および活性化状態と呼ばせていただきます。YFPとCFPの順番を変えるても大きな問題は起きないと考えていましたが、そうではないとする研究もあります文献33。また、YFP、CFP、センサー領域、リガンド領域の順列組み合わせは、ほかにもたくさんありますが、第一選択はここに挙げた順番です。以下にはさらに細かい点を解説します。
YFPのC末の長さ:
GFPのC末端11アミノ酸は、結晶解析でも構造決定できない自由運動を取る領域とされています。この領域をどれくらい残すかで、YFPとセンサー領域・リガンド領域との位置関係が規定されます。簡単には、全部残すと、かなり自由に動く領域になりますし、11個を全て削ると、YFPとセンサー・リガンドとがかっちり固定されるというイメージです。FRET効率は、CFPとYFPとの間の距離以外に、CFPの蛍光モーメントとYFPの励起モーメントの方向がどれくらい一致しているかが効きますので、YFPのC末端のアミノ酸1個のあるなしで、モーメントの向きで変化して、その結果FRET効率が劇的に変化することもあると報告されています。しかし、われわれの経験では今のところ、この領域が非常に大きくFRET効率を変化させたという経験は無く、現在は標準で11個削ったものを用いています。なお、N末側はドナーもアクセプターも最初のMet以外は削れません。
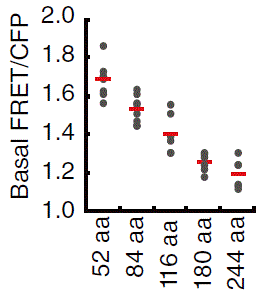
リンカー長 vs FRET/CFP比
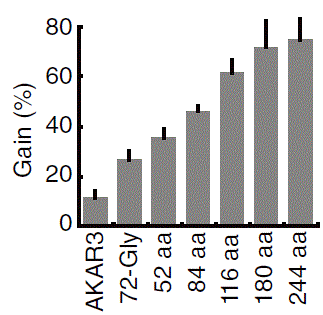
リンカー長 vs ゲイン
スペーサー:
おのおのの領域の間にはスペーサーと呼ばれるゼロから数十アミノ酸の長さの配列が挿入されています。私達の研究室ではスペーサーとしては、可動性を保つようにGly-Gly-Ser-Alaの繰り返し配列を用いたものをよく用いています。当初、それほど重要と思っていませんでした。30アミノ酸程度まで伸ばしてもあまり効果が無かったからです。また、昔のDNA合成ではそれほど長いリンカーを作ることもできませんでした。その後、センサー領域とリガンド領域の間のリンカーを100アミノ酸以上の長さにしたところ、非活性化状態のFRETが著名に低下し、結果としてシグナルノイズ比が顕著に上昇することを見出しました。現在、EVリンカーと命名した116アミノ酸のリンカーが研究室の標準となっています(文献35)。
PHOGEMONの局在:
PHOGEMONを細胞内のどこに最も多く発現させるべきかは非常に重要な問題です。この問題は重要なので別項目として詳述します。
PHOGEMON開発のためのアッセー系:
これから紹介するアッセー法は研究室で使用されてきた年代順に記載してあります。2017年現在、当研究室でルーチンに使用されている方法は、顕微鏡の分光機能を利用する方法です。
精製タンパク質を使う方法
大腸菌でプローブを産生させ、精製品を手にいれたのちに、その光学特性を調べる方法です。この方法は精製標品を用いて解析できますので、プローブの特性を十二分に調べることができます。生物物理の研究に用いるのであればこれは必須でしょう。しかし、もし、培養細胞で使うのが目的であるのなら、この戦術は間違っています。なぜなら、GFPが二つに加え、センサー部、リガンド部があるような大きなタンパク質を大腸菌で作らせると、殆ど不溶性になるからです。「組換えタンパク質が不溶性になるときはーーー」と、How to本にはいろいろ書いてありますが、わたし(MM)はほとんど失敗しました。蛇足ですが、私たちの研究室がこの分野である程度の成功を収めた最大の理由は、大腸菌でプローブを作ることを諦めたからです。2001年以前、FRETプローブは生物物理や化学の研究室で開発されており、そこでは大腸菌で作成した精製品を使うことが前提でした。培養細胞で発現させたFRETプローブでは、十分なプローブの特性の検討はできず、そのため、我々の戦略は、多くの先行グループから攻撃を受けました。しかし、2001年以降に開発されたプローブのほとんどは培養細胞で発現を確認しています。光学的な解析が十分できなくとも、生物学的プローブとしての性質が十分解析されれば問題ないと多くの研究者が考えるようになったからだと思います。しかし、最近はまた構造解析の分野などで組み換えタンパク質を作る技術が進み、大腸菌で精製したプローブを詳細に解析する研究も盛り返し始めています。
哺乳類培養細胞を使う方法
大腸菌に代わる発現系として、293細胞(ヒト腎臓由来)を用いる方法が開発されました。細胞由来の自家蛍光がありますから、厳密な光学的あるいは生化学的アッセーはできませんが、とにかく早く発現させて、光っているか、FRETが起きているかを簡便に調べることができます。ただし293細胞内では発現される組換えタンパク質はかなり多いとはいっても、蛍光分光光度計できれいなデータがとれるかどうかのぎりぎりぐらいです。できるだけ大量に産生するベクターが望ましいので、SV40複製基点を有する高発現ベクターを使うべきです。我々は、阪大の宮崎教授が開発し、最強という評判を得ているpCAGGSを長年使わせていただいています(文献3)。なお、実験のデザインにおいては、293細胞は試験管と割り切る気持ちが大切です。細胞内局在シグナルの重要性は先に述べましたが、これらはタンパク質の発現量を減らす傾向があります。モニターの開発段階では、生理的局在を無視して開発を進め、プローブのOn/OffがFRETではっきり検出できるものを作ることを優先し、それが完成した時点で、局在シグナルをつけるようにした方がいいでしょう。もともとは、293T細胞を可溶化して、それを分光光度計を使って解析していました。その後、293F細胞が使う方法が開発されました。この細胞は浮遊系で増やすことができる、タンパク質をたくさん産生できる、などの利点があります。さらに、この細胞の培地(Free style)は自家蛍光が低いので、細胞の懸濁液をそのまま、蛍光分光光度計のキュベットにいれるだけで蛍光特性がとれます。最近では、顕微鏡を使って蛍光プロフィールが取れるようになりましたので、トランスフェクション効率の高いHeLa細胞などを使うようになっています。
蛍光プロフィールのとり方
大腸菌等で精製品が手に入った場合の試験管内での実験法はあまり経験がないので省略し、以下は哺乳類細胞に発現させたプローブの蛍光プロフィールの取りかたを紹介します。
共焦点レーザ顕微鏡で取得した蛍光プロフィール
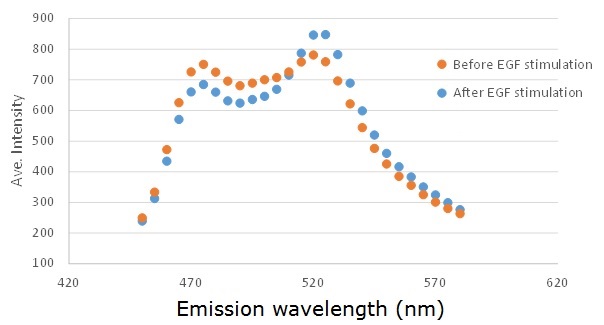
共焦点レーザ顕微鏡の分光機能を使う方法:
- 哺乳類細胞用の発現プラスミドを作る。
- ガラス底培養皿に播いたHeLa細胞にトランスフェクトする。
- 24から48時間後に倒立型共焦点レーザ顕微鏡の分光機能を使って、細胞の蛍光スペクトルを測定する。
- 倒立型蛍光タイムラプス顕微鏡にてタイムラプス観察を行い、適当な刺激をいれて、FRET効率が変化するかを調べる。
HeLa細胞等のトランスフェクション効率のよい細胞を使います。また、発現ベクターはpCAGGSなどできるだけ強力なプロモータをもつものが必要です。細胞由来の自家蛍光より十分明るい必要があるからです。35 mmカバーガラス底培養皿にHeLa細胞を2x10^5 cells/mLに播きます。そこに2 μgの発現プラスミドをトランスフェクトします。24-48時間後に、Fluobrite等の自家蛍光の低い培地(あるいはHBS, PBSなど)に置換し、共焦点レーザ顕微鏡で観察します。十分明るい細胞の蛍光プロフィールをを共焦点レーザ顕微鏡の分光機能を使って得ます。なお、十分なシグナルを得るために、ピンホールをほぼ全開にすることがきれいな蛍光プロフィールをコツです。
測定条件: EKAREV発現HeLa細胞 (EGF刺激前後); Olympus FV1000; Pixel:20.0; Size: 256x256; Zoom:x1; Laser:405nm 20.0%; HV:650V; LambdaScan:450-590nm; StepSize:5nm; Band Width:10nm
蛍光分光光度計と293F細胞を使う方法:
Raichu-Rasを例にとり解説します。6 wellプレート (浮遊細胞用)に293F細胞を1x10^6 cells/mLで1.5 mLずつ播きます。293F を浮遊状態で培養するために、培養皿は表面無処理のもの(浮遊細胞用)を使い、細胞はシェーカーで攪拌しながら培養します。そこに2 μgのpRaichuを、293Fectin (Invitrogen) によってプロトコルどおりにトランスフェクトします。できるだけタンパク質の発現量を多くするために36時間くらい CO2 インキュベーター内で細胞を培養します。細胞懸濁液を石英セルに移して、スターラーにより細胞を攪拌しながら、蛍光分光光度計で433 nm の光を当てて蛍光スペクトルを得ます。なお、FreeStyle(Invitrogen)は蛍光が少ないので、培地交換せずに測定することも可能です。
図6.1.1
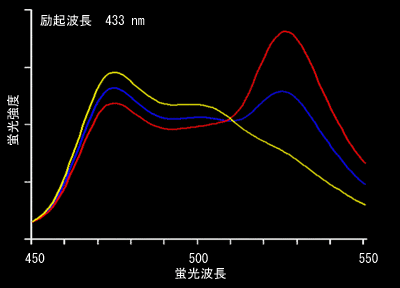
蛍光分光光度計と293T細胞を使う方法(実験例1):
10 cm培養皿に293T細胞をほぼいっぱいに増やし、10 μgの発現プラスミドをトランスフェクトします。できるだけタンパク質の発現量を多くするために48時間後くらいにアッセーをします。細胞を溶かす前に、正立の蛍光顕微鏡下で、トランスフェクション効率を確認しています。この際の細胞の観察に用いる蛍光フィルターセットは、FITCのものでかまいません。Triton-X100などの蛍光の少ないdetergentを含む緩衝液で可溶化して上清をとり、蛍光分光光度計を用いて433nmの励起光を当てて、蛍光スペクトルを得ます。代表的なデータを示します。図6.1.1)。なお、トランスフェクションしていないサンプルを調整し、自家蛍光のバックグランドファイルを作成しておくと、きれいなデータが作れます。このバックグランドファイルは原点がゼロになるように係数を掛けてから引くのがきれいなデータをとるためのこつです。
プロテアーゼを使ったFRETの確認: 530 nm付近のピークがFRETにより励起されたYFPからの蛍光であることを確認するために、すなわち、YFPが433nmの光で直接に励起されたものではないことを証明するために、Proteinase K処理することがあります。これは、GFPタンパク質がProteinase Kに対して抵抗性であることを利用し、介在するセンサー領域、リガンド領域、スペーサーなどの部分を消化して、YFPとCFPとに完全に分けてしまう方法です。上記293T細胞を用いる方法において、測定が終わった1.5 mlの液をマイクロチューブにとり、100 μg程度のProteinase Kとともに37度で30分ほど処理します。あるいは培養用のTrypsinを10 μl程度入れても十分です。 この条件で、YFPのピークが消失し、CFPのみの蛍光が観察されれば前に見えていた530nmの蛍光はFRETだった、という証明になります(黄色が分解後のサンプル)。
フローサイトメトリー/FACS
FRETバイオセンサーの安定発現が可能になったので、高発現細胞株を選抜するためにFACSをルーチンで使うようになりました。また、トランスジェニックマウスを使って細胞ごとのFRETバイオセンサーの発現量を比較する研究にも用いています(文献46)。CFPとYFPとを使ったFRET解析には、下記の光源とフィルターセットが必要です。
FACS
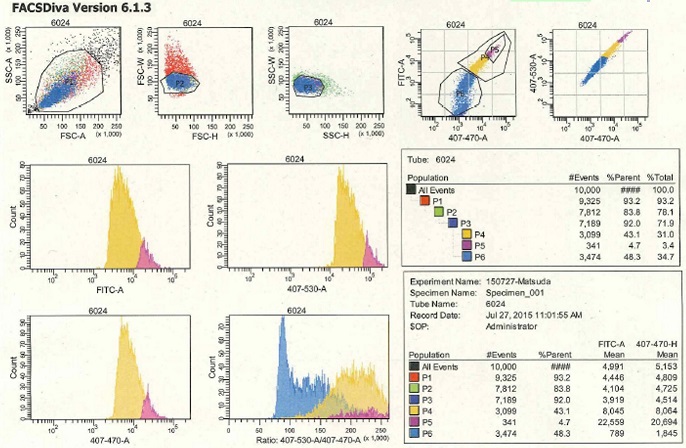
- 407nmの励起光を発する固形レーザーはオプションですが、CFPを励起するためには必須です。
- CFPの蛍光を観察するのに適したBand path filter(470AF40、Omega Optical, Inc.)を用いるとCFP由来の蛍光を効率よく検出できます。費用対効果の高い改良なので強くお勧めします。 FACSAriaのBand path filterは容易に交換できるので、例えば中央機器室においてあるような機械でも、自分たちが用いる蛍光物質のプロフィールを参照し、最も適したfilterを選ぶことでS/N比をあげることができます。
- 色素名の変更: 標準のFACSAriaのチャネルには蛍光色素の名前が付けられていますが、我々はこれらのチャネルの名は“励起波長”-“測定波長”という名に変えています (例、 407-470)。この設定はInstrument configuration で簡単に行えます。これにより、自分たちが用いている蛍光たんぱく質、蛍光色素の最適な励起波長と蛍光波長を調べることにより、誰がどのような蛍光物質を用いても、即座に理解し対応できます。
- FRET解析用ソフト: 我々はFRETのデータを標準添付のDiva softwareに加えWinList (Verity Software House, Inc.) を用いて解析しています。Diva softwareはFACSAriaのコントロールとデータ解析が行えるソフトなのだが、①生データをExcelに転送することができない、②Scatter plotのパラメータに、複雑な計算式で処理したデータを加えることができない、の二つの欠点があります。FRETを定量するためにはいくつかの補正が必要なので、WinListあるいはそれに替わるソフトは必須です。
- 実際の解析: BD社からアプリケーションマニュアルが配布されていますので、お問い合わせください。
観察方法: 細胞のイメージングに使う道具
図4.3.4
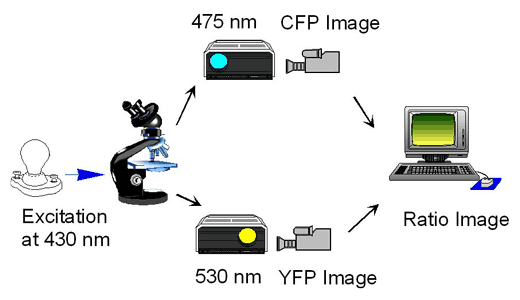
概要:
いよいよイメージングです。イメージングの方法を概念的に示しますと(図7.1.3)、細胞内のCFPを励起し、CFPの蛍光像とYFPの蛍光像とをCCDカメラで撮影し、コンピューターに保存し、最後に、CFPとYFPとの蛍光強度比に疑似カラーをつけて画像化するという流れです。培養細胞はリサーチ倒立型顕微鏡、器官培養は多光子インキュベータ蛍光顕微鏡、マウスの生体イメージングは正立型および倒立型多光子顕微鏡を使用しています。京都大学蛍光生体イメージング室の機材を参考にしてください。
図4.3.5

顕微鏡:
落射型蛍光顕微鏡の仕組みを理解することは、本プロジェクトには必須です(文献5)。顕微鏡はどこのメーカーでもいいと思いますが、赤外レーザを使ったオートフォーカス機能は今や必須です。タイムラプスイメージングの途中に焦点を合わせなおすのは、褪色の危険があり、すすめられません。また、XY軸モータードライブも必須です。この二つがあれば、複数の視野を同時にタイムラプスできます。数日にわたるタイムラプス実験の場合、50に近い視野をタイムラプスするということもやれます。ステージは、一般的な丸穴型ではなくて、96 wellにも対応できるものがあとあと役に立ちます。大は小を兼ねるといいますが、穴に関しては、大きい穴は小さい穴を兼ねる、というところです。また、ヒーター付きチャンバーをつけることも最初から考えておくべきです。さらに色収差などレンズに関する一般的光学特性を知っておくことも必要です。例えば当研究室ではCFPの像とYFPの像とで焦点が同時には合わないという事件がありました。高級レンズを購入していたのですが、色収差が十分に補正されていなかったらしいのです。レンズにロット差があるということを初めて知りました。
光源:
LED光源を第一選択としています。寿命がほぼ無限であること、光源がすぐに安定すること、光量を自在にコントロールでき、光量の記録を残しやすいこと、照明むらが少ないことなどが主な理由です。Arc lampを使うなら、広い波長で安定した光量を出すキセノンランプを使用するほうが融通が利きますが、寿命がかなり短いのが問題です。CFPとYFPのFRETのみの実験でしたら、安価な水銀ランプでも十分使用可能です。ただし、水銀ランプの波長は435 nmに大きなピークがあることを忘れてはいけません。長寿命のメタルハライドランプも使っています。最後に光源に関してもっとも大切なこと: YFPはとても褪色しやすいです。YFPが褪色するとFRETが減少して正しいデータがとれません。通常、光量は10%以下にして細胞を観察し、細胞を探す時間は必要最小限にすることを肝に銘じてください。 水銀ランプのときには、強力なNDフィルターが必要です。NDをはずさないと光が足りないような条件では、FRETの観察はほとんど無理です。「励起光は最小に当てる、蛍光は最大に回収する」これがFRET観察のコツです。
位相差・微分干渉:
細胞の形態を透過像で観察したい場合は、位相差か微分干渉等の屈折率を強度変換するシステムが必要です。位相差レンズは原理的に蛍光をロスするので、通常は推薦されませんが、実際には、許容範囲です。微分干渉(DIC)法において、干渉フィルターは蛍光(Emission)フィルターチェンジャーに入れておきますと、蛍光のロスは全くなくなり、FRET観察を行いながら細胞の形態も追跡できます。しかし、この蛍光フィルターチェンジャーにDICのフィルターを入れる方法は、DIC像の画質は本来の対物レンズ直下に入れた場合よりもかなり劣ります。もし、どうしてもDICの像もきれいにとりたい場合は、レンズ直下のキューブにDICフィルタを入れれば格段にきれいに取れます。この方法の弱点は、キューブの交換が1秒くらいかかるので、速い写真がとれないことです。
チャンバーおよび保温箱:
細胞を長時間観察するために、温度コントロールのできるチャンバーが必要です。昔は、フォーカスがずれるのを防ぐために、顕微鏡全体を覆うような大型のものを使っていましたが、オートフォーカス装置がほぼ標準となった現在では、96 wellにも対応できるCO2チャンバーを使っています。
CCDカメラ、フィルターチェンジャー、フィルターセット:
カメラの状況もここ数年ですっかり変わってしまいました。CCDカメラはほぼsCMOSカメラに駆逐されつつあると言ってもいいでしょう。当研究室は”もったいない”の精神から古いCCDを使ってますので、sCMOSについてあまり申し上げることはありません。フィルターチェンジャーは6連のを使っています。フィルターセットはOmega、Chroma、Semrock、朝日分光の四社から購入しています。値段が結構違うので、適宜選択して賢い買い物をするように心がけます。朝日分光とSemlockの製品はハードコートなので、錆びにくく湿気の多い日本に向いています。フィルターは錆びるというのは覚えておく必要があります。年に1回はフィルターチェンジャーを開けて覗いてみてください(機械音痴の人でも、それくらいはやってください)。蛇足ですが買ってきたフィルターの特性を分光光度計で調べてみるのは勉強になります。分光光度計のキュベットを取り外して、光路に直交するように置けば簡単に測定できます。ダイクロイックミラーだけは、45度に傾けて置くのを忘れずに。参考までに我々の顕微鏡の一つに入っているミラーセットを示します(表7.9.1)。最後にフィルターに関してもっとも大事なこと:素手でさわらない。つけてしまった指紋をこすってとってはならない。ミラーは精密機械です。

画面分割装置:
これまでに紹介したシステムでは、430 nmで励起して、まず475 nmのCFPの蛍光画像をとり、次に蛍光フィルターを替えて530 nmのYFPの蛍光画像を取るものです。このようなratiometryのみを行う方のために、1回の撮影でCFPとYFPの両方の画像を撮影するシステムがハマホトとRoperの両社からを販売されています。 Flow Chamberを使って血球の細胞を見る時のように早い動きの細胞を観察するために使っています。新しいW-View GEMINIは小さくなって、視野が大きくなりました。
利点
- CFPとYFPのイメージを同時に取れるので、速い動きのイメージングに強い。
- フィルターを交換することによるCFPとYFPの画像のずれ(misregistration)を押さえることができる。
- 励起が1回で済むので、褪色が少ない。
欠点
- 透過像が同時にはとれない。
- 画面が半分の大きさしかない。
- カメラとポートが占有されてしまう。
培地:
培地はフェノールレッドの入っていないものを使う必要があります。イメージング用にFluoBriteが発売されています。初代神経細胞などは特に、光に弱いので、B27等のサプリメントを足します。また、血清も蛍光を発しますので、無血清でできればそのほうがいいですが、どうしても細胞が傷む場合は加えています。その他サプリメントを加える場合は自家蛍光がないかを確認してください。なお、増殖因子を加えるための培地はfreshなものではなくて、実験に使う細胞から前もって0.5 ml程度抜いておき、それを用います。培地が変わるだけでシグナルが入る場合がしばしばあります。
データ解析ソフト:
当研究室ではMolecular Device社のMetaMorphを使っています。研究室で購入すると高価なソフトではありますが、京大内ではネットワークライセンスがあるので誰でも使用できます。FIJI (Image-J)は無料が魅力で、機能も優れています。しかし、同じようなことができるコマンドが多数あるので、どのコマンドを使うべきかがわかりにくいのが問題です。なお、多数の画像解析にはマクロを作り、処理を自動化することが必須です。マクロ作成に関してはMetaMorphのほうFIJIよりは容易といえるでしょう。さらに詳細に画像を解析したり、モデルを作ったりということになるとMATLABが優れています。京大ではアカデミックユーザーグループがあり、非常に安く使うことができます。生物系研究者にはハードルが高いですが、若い院生諸君にはぜひチャレンジして欲しいと思います。膨大な画像データの保管は、多光子顕微鏡の研究が始まって以来、ますます頭を痛めている問題ですが、いまだ解決できていません。
その他、特殊な装置:
共焦点レーザー顕微鏡: 半導体レーザーが安価になり、CFP励起もさほど問題ではなくなりました。スペクトル解析に対応した顕微鏡も標準になりつつあり、これからはこちらが標準になるかもしれません。
全反射型顕微鏡: エバネッセント場顕微鏡も市販品が出て、容易にできるようになりました。
2光子レーザー顕微鏡:(高価な割には戦力にならないので)戦艦大和とあだ名がつけられていました。しかし、in vivo imagingが盛んになってきたのに伴い、いまや主戦力として活躍している顕微鏡です。京都大学生体イメージング室には共用の多光子顕微鏡が5台ありますが、予約がなかなか取れないのが問題になっています。世界でもっとも稼働率が高いという噂です。
研究の具体例の紹介 (PHOGEMON 各論)
Eevee:
Eeveeシリーズは、タンパク質リン酸化酵素の基質配列のリン酸化を定量することにより、目的とするリン酸化酵素の活性化を測定するFRETバイオセンサー群です(文献34)。多数の安定発現培養細胞株やトランスジェニックマウス(文献35)が作成されており、タンパク質リン酸化酵素が生体内でどのように制御されているかを解析するための貴重なツールとなています。特にタンパク質リン酸化酵素ERKは細胞増殖のみならず、神経分化や細胞遊走など、細胞のアクティビティと極めてよく相関するため、組織内での細胞の状態をモニターするのに便利です。研究室では、細胞の活性化状態が周囲の細胞に伝搬することを培養細胞のみならず生きたマウスの組織でも明らかにしました。これは、FRETバイオセンサーを使って初めて分かった現象であり、生体内には我々が見たことがない様々な現象があり、FRETバイオセンサーがそれを明らかにする非常によいツールであることを示しています。下記は代表的なバイオセンサーとそれをつかった論文です。
ERK MAPキナーゼの自発的発火と伝搬 (文献36)
生体表皮における新規ERK活性化現象(SPREAD) (文献37); Video image
血管から遊走する好中球におけるERKとAキナーゼの活性制御 (文献38)Video image
腫瘍血管新生におけるAキナーゼの活性制御 (文献39)
腫瘍組織内でのストレス反応性リン酸化酵素Tak1の活性変化 (文献41)
高感度Aktバイオセンサー (文献43)
Kiss technologyで作成したROCKバイオセンサー (文献44)
Raichu:
Ras and interacting protein chimeric unitの略。RasスーパーファミリーGタンパク質の活性化モニターの総称です(文献12)。
Raichuについての詳細な記述はここをご覧ください。
上皮細胞増殖因子によるRasの活性化 (文献4)
図5.1.1
Rasはヒトの癌の30%に活性化型変異が認められる癌遺伝子の王様である。この分子は低分子量GTP結合タンパク質の代表で、細胞内の分子スイッチとして機能している(図5.1.1)。すなわち通常はGDPに結合した非活性化型として存在するが、グアニンヌクレオチド交換因子の存在下にGDPがGTPに置換されると、構造変化が起こり、その中のエフェクター結合領域がエフェクターに結合し、情報をエフェクターに伝播する。つまり、GDPをGTPに置換することで活性化型に変化する。エフェクターは多数知られているが、もっとも有名なエフェクター分子はセリンスレオニンキナーゼ型癌遺伝子産物であるRafである。RafはRasと結合することにより活性化される。図5.1.1では、不活性化型を青で、活性化型を赤で示してある。GTP型のRasは、GTP水解促進酵素(GAP)の存在下にGTPをGDPに水解し、不活性化型に戻る。
さて、Rasを始めとして多くのGタンパク質は膜画分に存在することが知られている。しかし、膜画分といっても、細胞膜や、小胞体、Golgiなど、いろんな場所がある。Rasも大部分は細胞膜に存在するものの、Golgi装置なででは異なった機能を発揮しうるのではないかと最近、報告されている。したがって、さまざまな刺激に応じて、Rasがどこで活性化されるかを観察することが必須である。Raichu-Rasプローブはこれを初めて可能にしたものである。
cAMPによるeGRF発現細胞でのRasの活性化(文献19)
細胞運動時のRacとCdc42の活性化 (文献18)
細胞運動時のRhoの活性化(文献21)
細胞分裂時のRhoファミリーGタンパク質の活性変化(文献20)
エキソサイトーシス過程でのTC10タンパク質の活性化イメージング(文献22)
葉状突起でのRalAの活性化(文献30)
貪食作用のキー分子Rab5の働きを可視化(文献31)
RRas(文献25)
Picchu:
図5.2.1
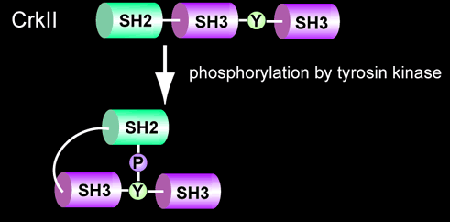
Crkは、アダプター分子群発見の先駆けとなった分子であり、SH2ドメインとリン酸化チロシンの結合によるチロシンリン酸化情報伝達機構の解明をもたらした分子である。1988年にBruce MayerはニワトリRNA型肉腫ウィルスCT10より癌遺伝子v-crkを同定した。CrkはSH2、SH3ドメインのみで構成される分子であり、それ自身は酵素活性を持たないが、リン酸化チロシンとSH2ドメインが結合することによってC3GやDOCK180等のSH3ドメイン結合分子を基質の存在する場所にリクルートするアダプタータンパク質質である。また、他のアダプター分子と異なりCrk自身もSrcファミリーチロシンキナーゼ、AblやEGF Receptor等の増殖因子レセプターによってチロシンリン酸化され、分子内SH2ドメイン-リン酸化チロシン結合による構造変化を起こします(図5.2.1)。 PicchuはPhosphorylation indicator of Crk chimeric unitの略。アダプター分子Crkのリン酸化状態をモニターするために作成されていますが(文献7)、原理的にはチロシンリン酸化酵素の活性化モニターです。プローブの詳細な記述はここをご覧ください。
上皮細胞増殖因子によるCrkリン酸化の可視化(文献7)
Pippi:
Phophatidylinositolphosphate indicatorの略。東大梅沢研究室のアイデアによるPIP3モニターの骨格を使っています(文献23)。Pippiは、リン脂質をモニターするプローブです。プローブの使用例はここをご覧ください。
Prin:
Photometric Raf indicatorの略(文献27, 28)。Prinは、Rafの構造変化を検出するプローブです。 プローブの使用例はここをご覧ください。
Miu2:
MAPK indicator unit ERK2の略(文献29)。Miu2は、ERK2の構造変化を検出するプローブです。このプローブで検出される構造変化はMEKとの結合によるものです。
Akind:
Akt indicatorの略(文献26)。Akindは、Aktの構造変化をモニターするプローブです。
困ったときは
- 何も刺激していないのにRatio(FRET/CFP)が下がってくる: Ratio(FRET/CFP)に関するトラブルのときは、まず、FRET、CFPの値がどう変化しているかを確認してください。ともに減少している場合は褪色しています。撮影間隔を上げるか、露光時間を短くしてください。CFPが上昇し、FRETが低下している場合は、理由は不明ですが、正しくFRETが変化しています。
- 何も刺激していないのにRatio(FRET/CFP)が上がってくる: ① 細胞を探すときに褪色してしまった。しばらく待つと安定することがあります。② タンパク質のフォールディングに時間がかかっている。この場合、YFPの蛍光もCFPの蛍光も増加してきます。トランスフェクション後にあまり時間がたっていないと時々おきます。③ 細胞の大きさが変わった。細胞の大きさが変わったりして、局所のプローブの量が変化すると、Ratioが高く、あるいは低くなることがあります。バックグランドの補正の問題です。
- 撮影中に焦点がずれる: 原因は様々です。熱膨張に起因するものを防ぐには、保温箱を十分暖めてから撮影してください。あるいは顕微鏡にクーラーの風が直接当たっていませんか?培養皿を固定する方法も工夫が必要です。オートフォーカスがついている場合は、ガラス面の汚れあるいはサンプル間の移動速度が速すぎるなどの原因が多いように思います。
- 細胞の片側で徐々にRatioが高く(低く)見えるが、いつも同じ向きだ: 励起光にむらがありませんか?芯出しをきっちりやってください。
- 細胞の辺縁でRatioが高く(低く)見える: misregistrationです。YFPとCFPの画像の位置ををソフト上で微調整してください。
- プローブを入れた細胞が元気がない: プローブによっては過剰発現で毒性をもつなどの影響がありえます。プローブの発現量の少ない細胞でやるしかないでしょう。
- 安定細胞株ができない: piggyBac, Tol2などのTransoposonを使う。レトロあるいはレンチウイルスを使う場合は、CFPとYFPの相同性を減らした変異体を使ってください(文献44)。
サポート
本研究は過去20年にわたり文部科学省をはじめ様々な財団の支援を受けて行われてきました。諸官庁ならびに本プロジェクトを支持していただいた審査委員の先生にこの場を借りてお礼申し上げます。
- 文部科学省科学研究費基盤B、基盤A
- 文部科学省特定研究「たんぱく可視化」
- 科学技術振興調整費「先端技術分野」
- 文部科学省がん特定研究「がん科学のニューフロンティア」
- 新学術領域研究「蛍光生体イメージング」
- 革新的細胞解析研究プログラム(セルイノベーション)
- 生命動態システム科学研究推進拠点事業
- 新学術領域研究領域「レゾナンスバイオ」
- 中谷医工計測技術振興財団
- CREST「光の特性を活用した生命機能の時空間制御技術の開発と応用」
謝辞、著作権
これまで開発したFRETプローブは研究室の学生ならびにスタッフたちの汗と涙の結晶であることを付記し、深甚なる感謝の意を表したいと思います。本文の著作権は、松田道行(京都大学大学院医学研究科病態生物医学)が有しています。